# Unfiltered human PBMCs (10X Genomics)
## Introduction
Here, we describe a brief analysis of the peripheral blood mononuclear cell (PBMC) dataset from 10X Genomics [@zheng2017massively].
The data are publicly available from the [10X Genomics website](https://support.10xgenomics.com/single-cell-gene-expression/datasets/2.1.0/pbmc4k),
from which we download the raw gene/barcode count matrices, i.e., before cell calling from the _CellRanger_ pipeline.
## Data loading
```r
library(DropletTestFiles)
raw.path <- getTestFile("tenx-2.1.0-pbmc4k/1.0.0/raw.tar.gz")
out.path <- file.path(tempdir(), "pbmc4k")
untar(raw.path, exdir=out.path)
library(DropletUtils)
fname <- file.path(out.path, "raw_gene_bc_matrices/GRCh38")
sce.pbmc <- read10xCounts(fname, col.names=TRUE)
```
```r
library(scater)
rownames(sce.pbmc) <- uniquifyFeatureNames(
rowData(sce.pbmc)$ID, rowData(sce.pbmc)$Symbol)
library(EnsDb.Hsapiens.v86)
location <- mapIds(EnsDb.Hsapiens.v86, keys=rowData(sce.pbmc)$ID,
column="SEQNAME", keytype="GENEID")
```
## Quality control
We perform cell detection using the `emptyDrops()` algorithm, as discussed in Section \@ref(qc-droplets).
```r
set.seed(100)
e.out <- emptyDrops(counts(sce.pbmc))
sce.pbmc <- sce.pbmc[,which(e.out$FDR <= 0.001)]
```
```r
unfiltered <- sce.pbmc
```
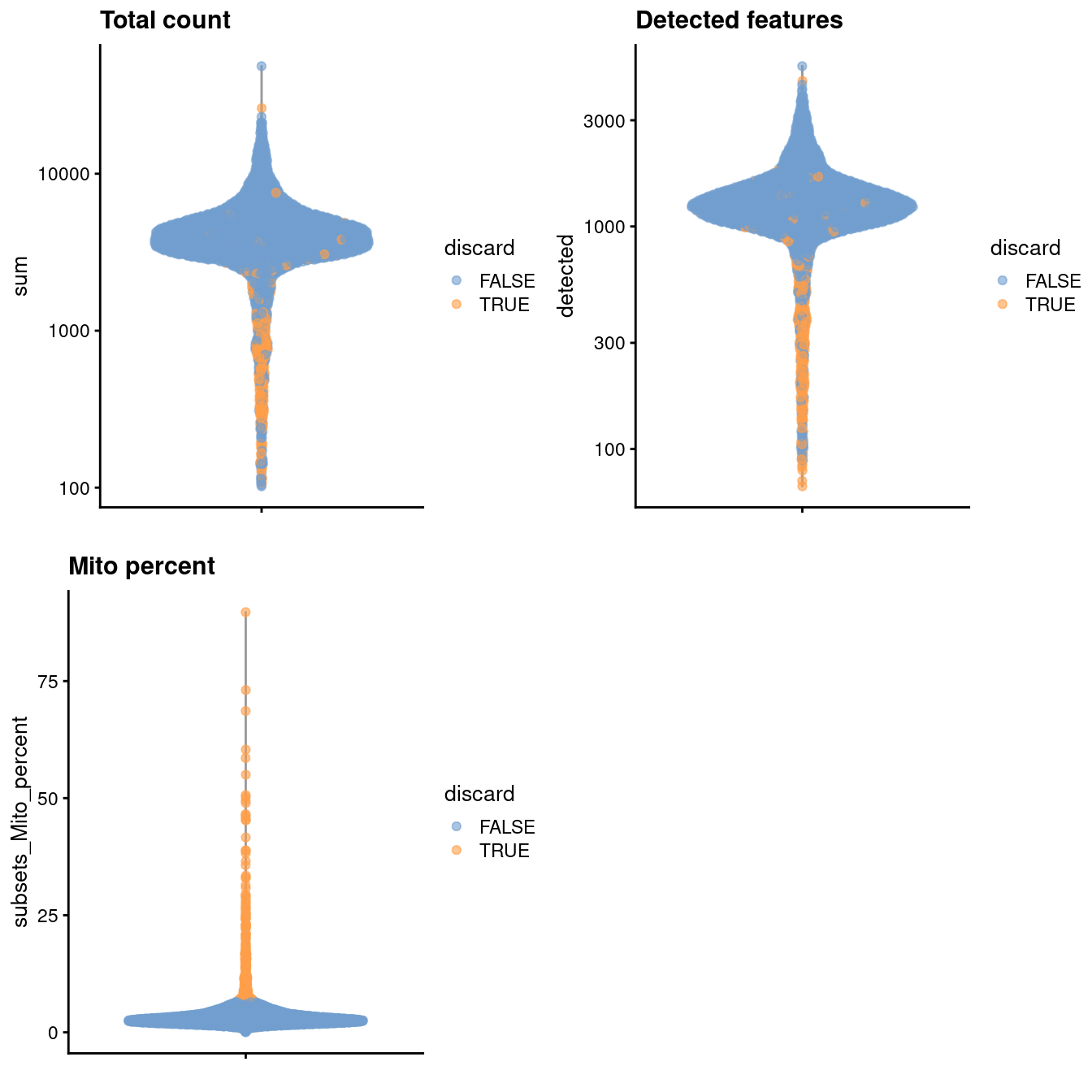
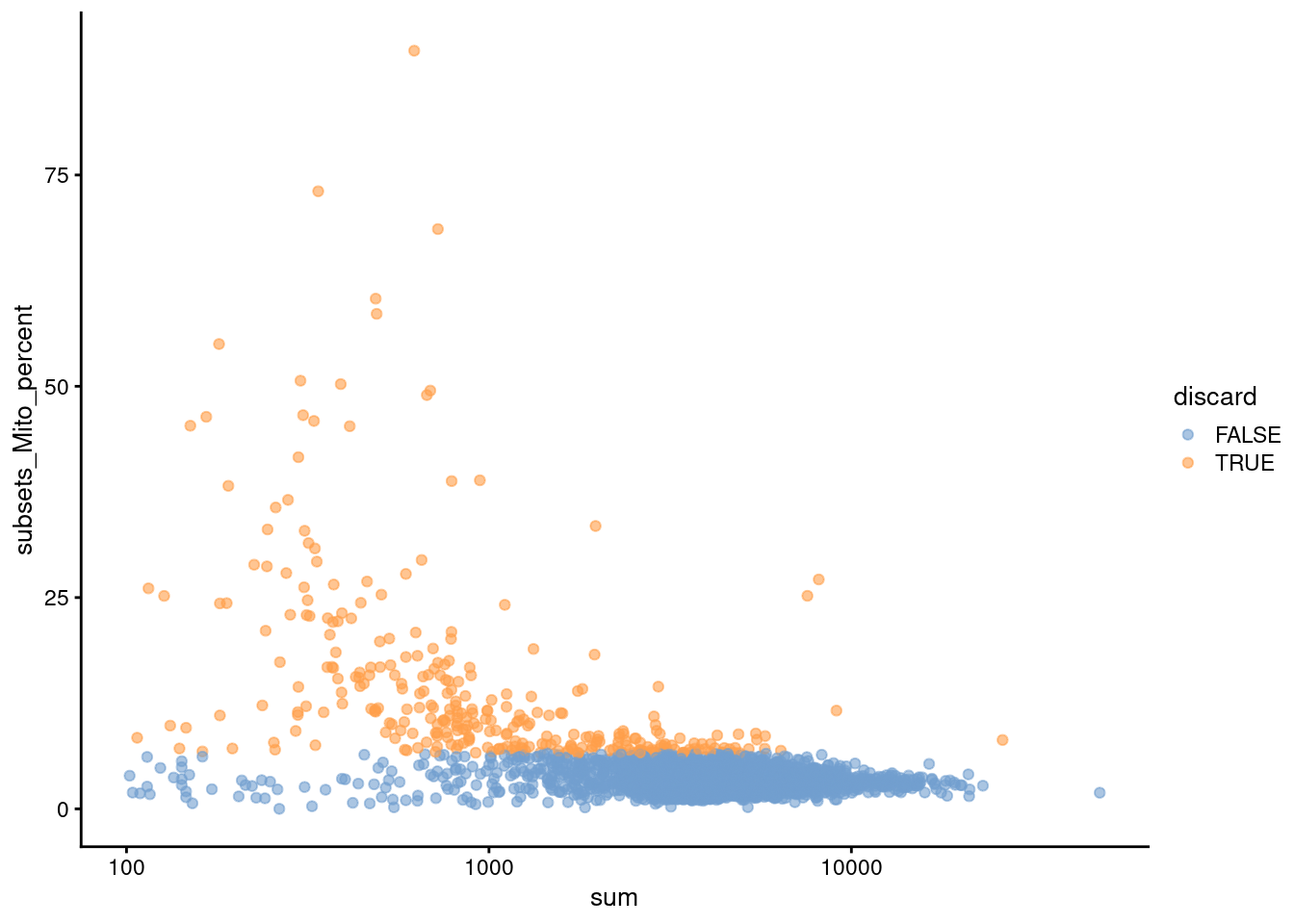
We use a relaxed QC strategy and only remove cells with large mitochondrial proportions, using it as a proxy for cell damage.
This reduces the risk of removing cell types with low RNA content, especially in a heterogeneous PBMC population with many different cell types.
```r
stats <- perCellQCMetrics(sce.pbmc, subsets=list(Mito=which(location=="MT")))
high.mito <- isOutlier(stats$subsets_Mito_percent, type="higher")
sce.pbmc <- sce.pbmc[,!high.mito]
```
```r
summary(high.mito)
```
```
## Mode FALSE TRUE
## logical 3985 315
```
```r
colData(unfiltered) <- cbind(colData(unfiltered), stats)
unfiltered$discard <- high.mito
gridExtra::grid.arrange(
plotColData(unfiltered, y="sum", colour_by="discard") +
scale_y_log10() + ggtitle("Total count"),
plotColData(unfiltered, y="detected", colour_by="discard") +
scale_y_log10() + ggtitle("Detected features"),
plotColData(unfiltered, y="subsets_Mito_percent",
colour_by="discard") + ggtitle("Mito percent"),
ncol=2
)
```
(\#fig:unref-unfiltered-pbmc-qc)Distribution of various QC metrics in the PBMC dataset after cell calling. Each point is a cell and is colored according to whether it was discarded by the mitochondrial filter.
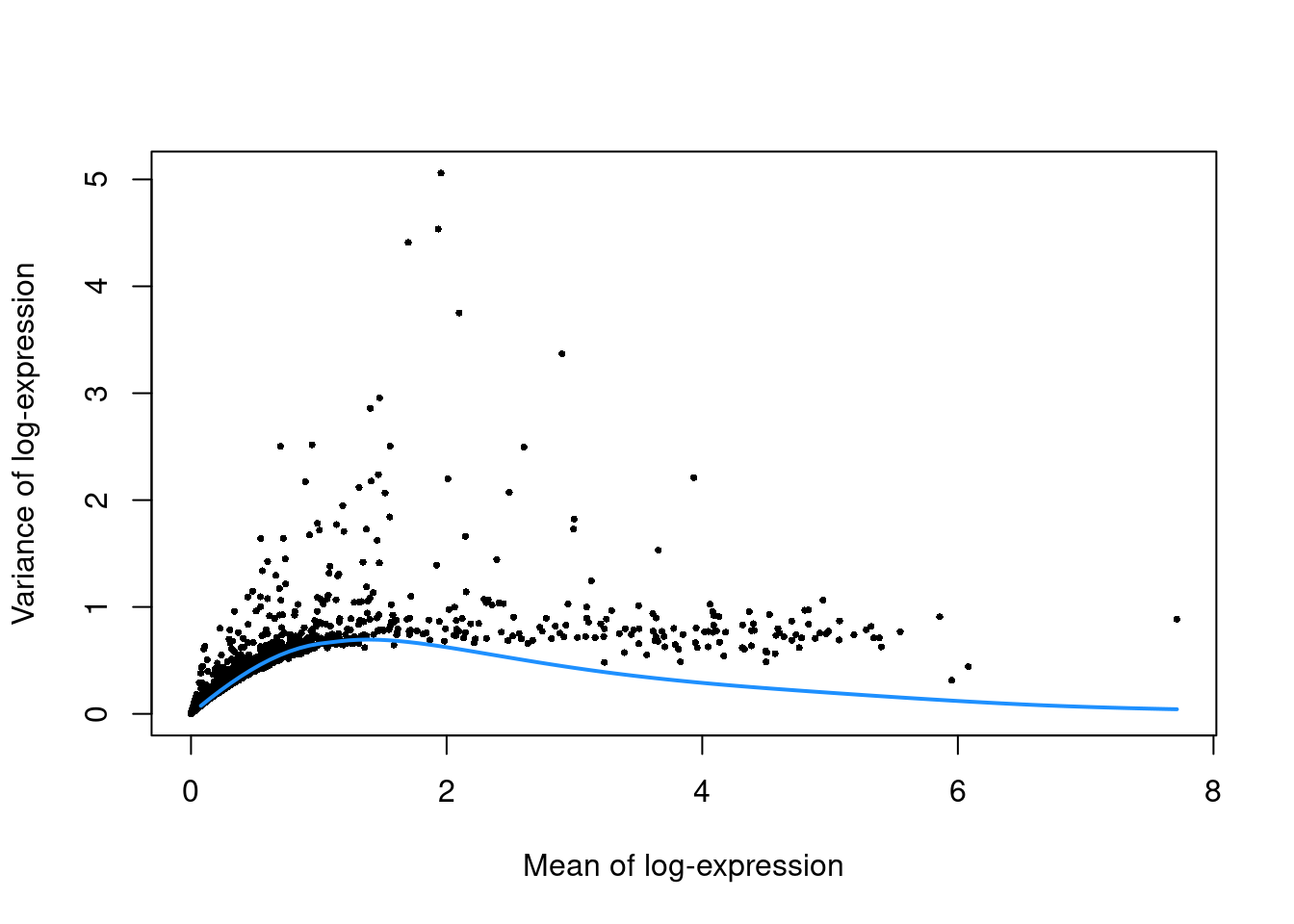
(\#fig:unref-unfiltered-pbmc-var)Per-gene variance as a function of the mean for the log-expression values in the PBMC dataset. Each point represents a gene (black) with the mean-variance trend (blue) fitted to simulated Poisson counts.
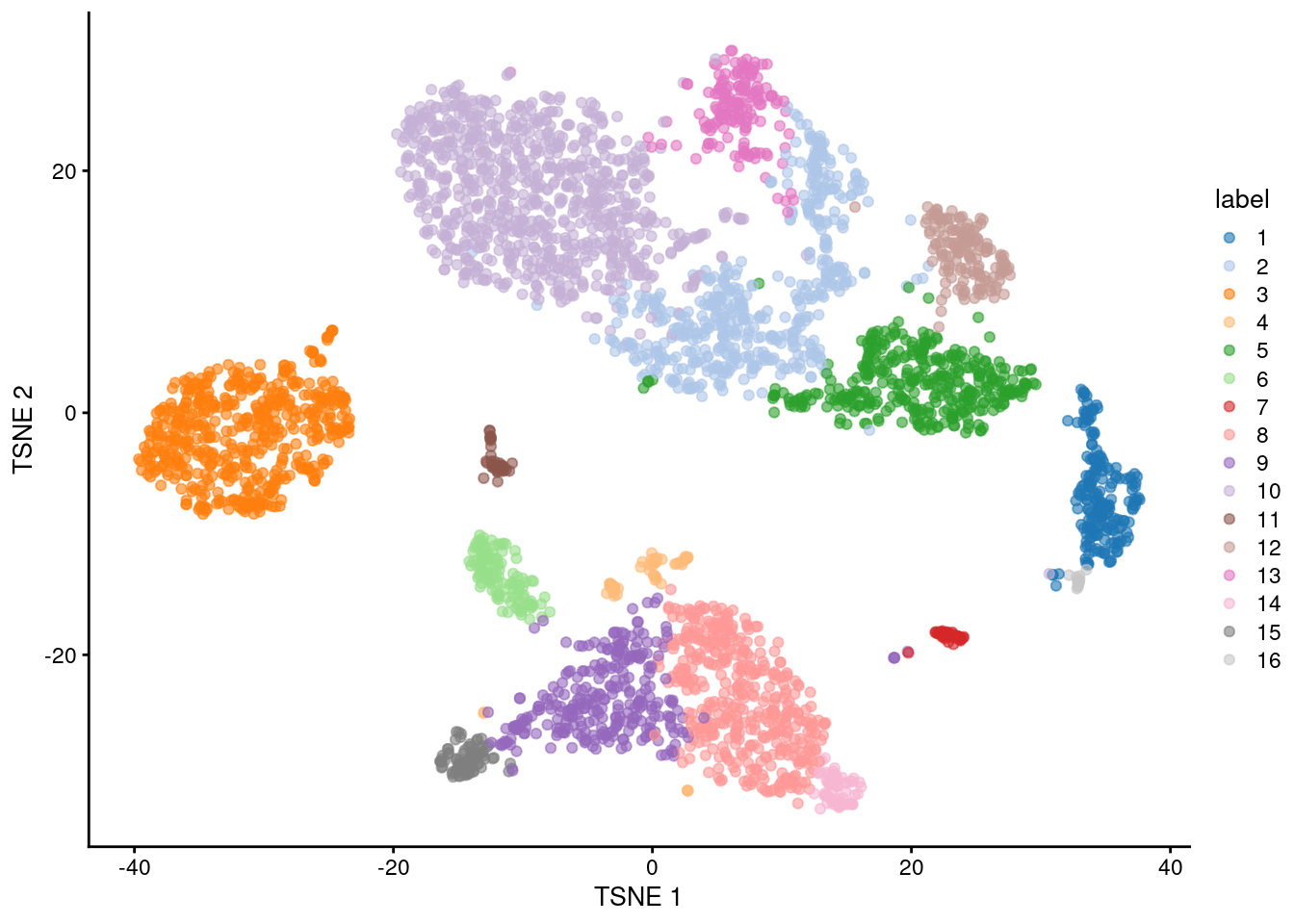
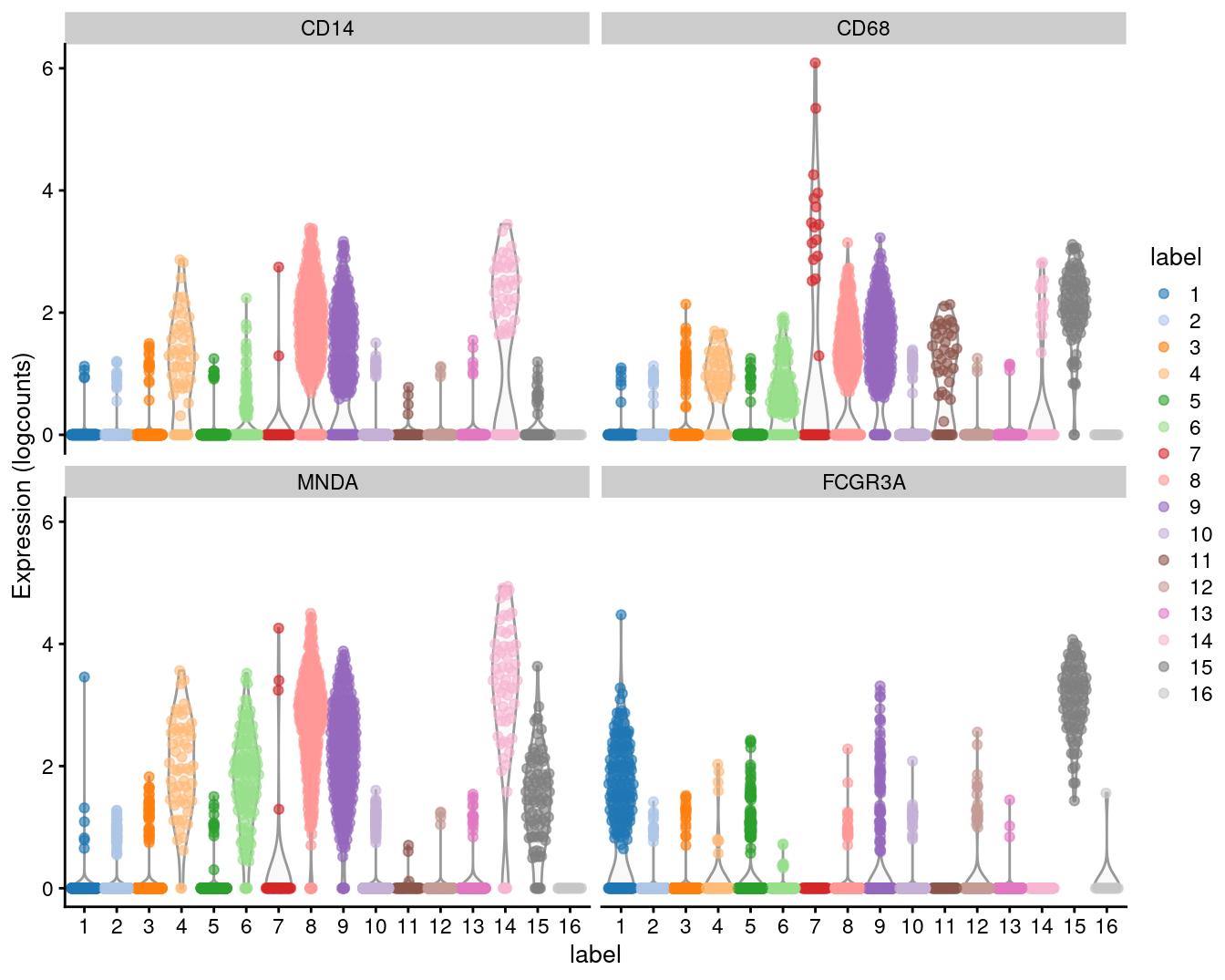
(\#fig:unref-unfiltered-pbmc-tsne)Obligatory $t$-SNE plot of the PBMC dataset, where each point represents a cell and is colored according to the assigned cluster.