---
output:
html_document
bibliography: ref.bib
---
# Integrating with protein abundance
## Motivation
Cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) is a technique that quantifies both gene expression and the abundance of selected surface proteins in each cell simultaneously [@stoeckius2017simultaneous].
In this approach, cells are first labelled with antibodies that have been conjugated to synthetic RNA tags.
A cell with a higher abundance of a target protein will be bound by more antibodies, causing more molecules of the corresponding antibody-derived tag (ADT) to be attached to that cell.
Cells are then separated into their own reaction chambers using droplet-based microfluidics [@zheng2017massively].
Both the ADTs and endogenous transcripts are reverse-transcribed and captured into a cDNA library; the abundance of each protein or expression of each gene is subsequently quantified by sequencing of each set of features.
This provides a powerful tool for interrogating aspects of the proteome (such as post-translational modifications) and other cellular features that would normally be invisible to transcriptomic studies.
How should the ADT data be incorporated into the analysis?
While we have counts for both ADTs and transcripts, there are fundamental differences in nature of the data that make it difficult to treat the former as additional features in the latter.
Most experiments involve only a small number of antibodies (<20) that are chosen by the researcher because they are of _a priori_ interest, in contrast to gene expression data that captures the entire transcriptome regardless of the study.
The coverage of the ADTs is also much deeper as they are sequenced separately from the transcripts, allowing the sequencing resources to be concentrated into a smaller number of features.
And, of course, the use of antibodies against protein targets involves consideration of separate biases compared to those observed for transcripts.
In this chapter, we will describe some strategies for integrated analysis of ADT and transcript data in CITE-seq experiments.
We will demonstrate using a PBMC dataset from 10X Genomics that contains quantified abundances for a number of interesting surface proteins.
We conveniently obtain the dataset using the *[DropletTestFiles](https://bioconductor.org/packages/3.12/DropletTestFiles)* package, after which we can create a `SingleCellExperiment` as shown below.
```r
library(DropletTestFiles)
path <- getTestFile("tenx-3.0.0-pbmc_10k_protein_v3/1.0.0/filtered.tar.gz")
dir <- tempfile()
untar(path, exdir=dir)
# Loading it in as a SingleCellExperiment object.
library(DropletUtils)
sce <- read10xCounts(file.path(dir, "filtered_feature_bc_matrix"))
sce
```
```
## class: SingleCellExperiment
## dim: 33555 7865
## metadata(1): Samples
## assays(1): counts
## rownames(33555): ENSG00000243485 ENSG00000237613 ... IgG1 IgG2b
## rowData names(3): ID Symbol Type
## colnames: NULL
## colData names(2): Sample Barcode
## reducedDimNames(0):
## altExpNames(0):
```
## Setting up the data
The `SingleCellExperiment` class provides the concept of an "alternative Experiment" to store data for different sets of features but the same cells.
This involves storing another `SummarizedExperiment` (or an instance of a subclass) _inside_ our `SingleCellExperiment` where the rows (features) can differ but the columns (cells) are the same.
In previous chapters, we were using the alternative Experiments to store spike-in data, but here we will use the concept to split off the ADT data.
This isolates the two sets of features to ensure that analyses on one set do not inadvertently use data from the other set, and vice versa.
```r
sce <- splitAltExps(sce, rowData(sce)$Type)
altExpNames(sce)
```
```
## [1] "Antibody Capture"
```
```r
altExp(sce) # Can be used like any other SingleCellExperiment.
```
```
## class: SingleCellExperiment
## dim: 17 7865
## metadata(1): Samples
## assays(1): counts
## rownames(17): CD3 CD4 ... IgG1 IgG2b
## rowData names(3): ID Symbol Type
## colnames: NULL
## colData names(0):
## reducedDimNames(0):
## altExpNames(0):
```
At this point, it is also helpful to coerce the sparse matrix for ADTs into a dense matrix.
The ADT counts are usually not sparse so storage as a sparse matrix provides no advantage; in fact, it actually increases memory usage and computational time as the indices of non-zero entries must be unnecessarily stored and processed.
From a practical perspective, this avoids unnecessary incompatibilities with downstream applications that do not accept sparse inputs.
```r
counts(altExp(sce)) <- as.matrix(counts(altExp(sce)))
counts(altExp(sce))[,1:10] # sneak peek
```
```
## [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10]
## CD3 18 30 18 18 5 21 34 48 4522 2910
## CD4 138 119 207 11 14 1014 324 1127 3479 2900
## CD8a 13 19 10 17 14 29 27 43 38 28
## CD14 491 472 1289 20 19 2428 1958 2189 55 41
## CD15 61 102 128 124 156 204 607 128 111 130
## CD16 17 155 72 1227 1873 148 676 75 44 37
## CD56 17 248 26 491 458 29 29 29 30 15
## CD19 3 3 8 5 4 7 15 4 6 6
## CD25 9 5 15 15 16 52 85 17 13 18
## CD45RA 110 125 5268 4743 4108 227 175 523 4044 1081
## CD45RO 74 156 28 28 21 492 517 316 26 43
## PD-1 9 9 20 25 28 16 26 16 28 16
## TIGIT 4 9 11 59 76 11 12 12 9 8
## CD127 7 8 12 16 17 15 11 10 231 179
## IgG2a 5 4 12 12 7 9 6 3 19 14
## IgG1 2 8 19 16 14 10 12 7 16 10
## IgG2b 3 3 6 4 9 8 50 2 8 2
```
## Quality control
As with the endogenous genes, we want to remove cells that have failed to capture/sequence the ADTs.
Recall that droplet-based libraries will contain contamination from ambient solution (Section \@ref(qc-droplets)), in this case containing containing conjugated antibodies that are either free in solution or bound to cell fragments.
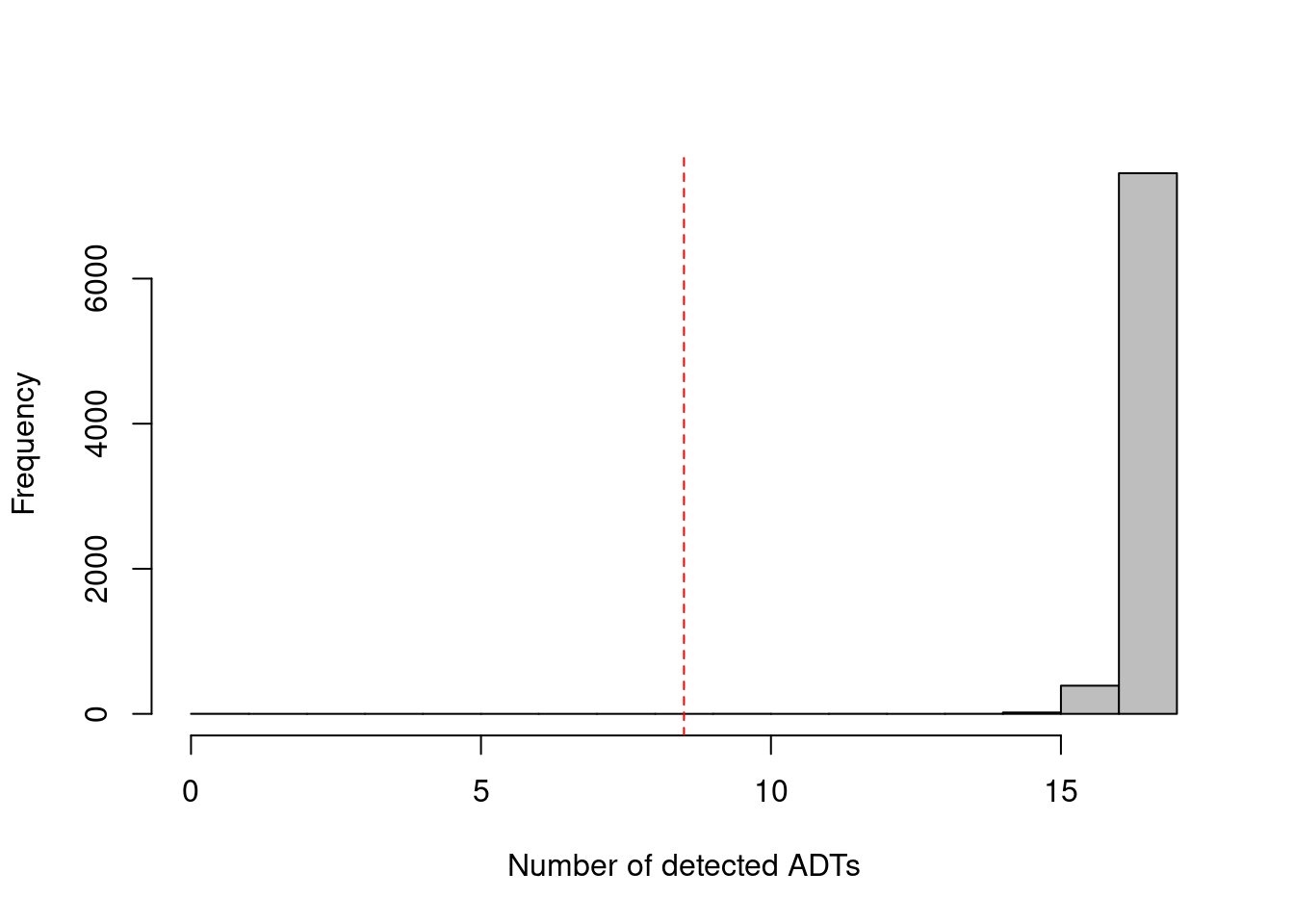
As the ADTs are (relatively) deeply sequenced, we can expect non-zero counts for most ADTs in each cell due to contamination (Figure \@ref(fig:cite-detected-ab-hist); if this is not the case, we might suspect some failure of ADT processing for that cell.
We thus remove cells that have unusually low numbers of detected ADTs, defined here as less than or equal to half of the total number of tags.
```r
# Applied on the alternative experiment containing the ADT counts:
library(scuttle)
df.ab <- perCellQCMetrics(altExp(sce))
n.nonzero <- sum(!rowAlls(counts(altExp(sce)), value=0L))
ab.discard <- df.ab$detected <= n.nonzero/2
summary(ab.discard)
```
```
## Mode FALSE TRUE
## logical 7864 1
```
```r
hist(df.ab$detected, col='grey', main="", xlab="Number of detected ADTs")
abline(v=n.nonzero/2, col="red", lty=2)
```
(\#fig:cite-detected-ab-hist)Distribution of the number of detected ADTs across all cells in the PBMC dataset. The red dotted line indicates the threshold below which cells were removed.
To elaborate on the filter above: we use `n.nonzero` to exclude ADTs with zero counts across all cells, which avoids inflation of the total number of ADTs due to barcodes that were erroniously included in the annotation.
The use of the specific threshold of 50% aims to avoid zero size factors during median-based normalization (Section \@ref(cite-seq-median-norm)), though it is straightforward to use more stringent thresholds if so desired.
We do not rely only on `isOutlier()` as the MAD is often zero in deeply sequenced datasets - where most cells contain non-zero counts for all ADTs - such that filtering would discard useful cells that only detect _almost_ all of the ADTs.
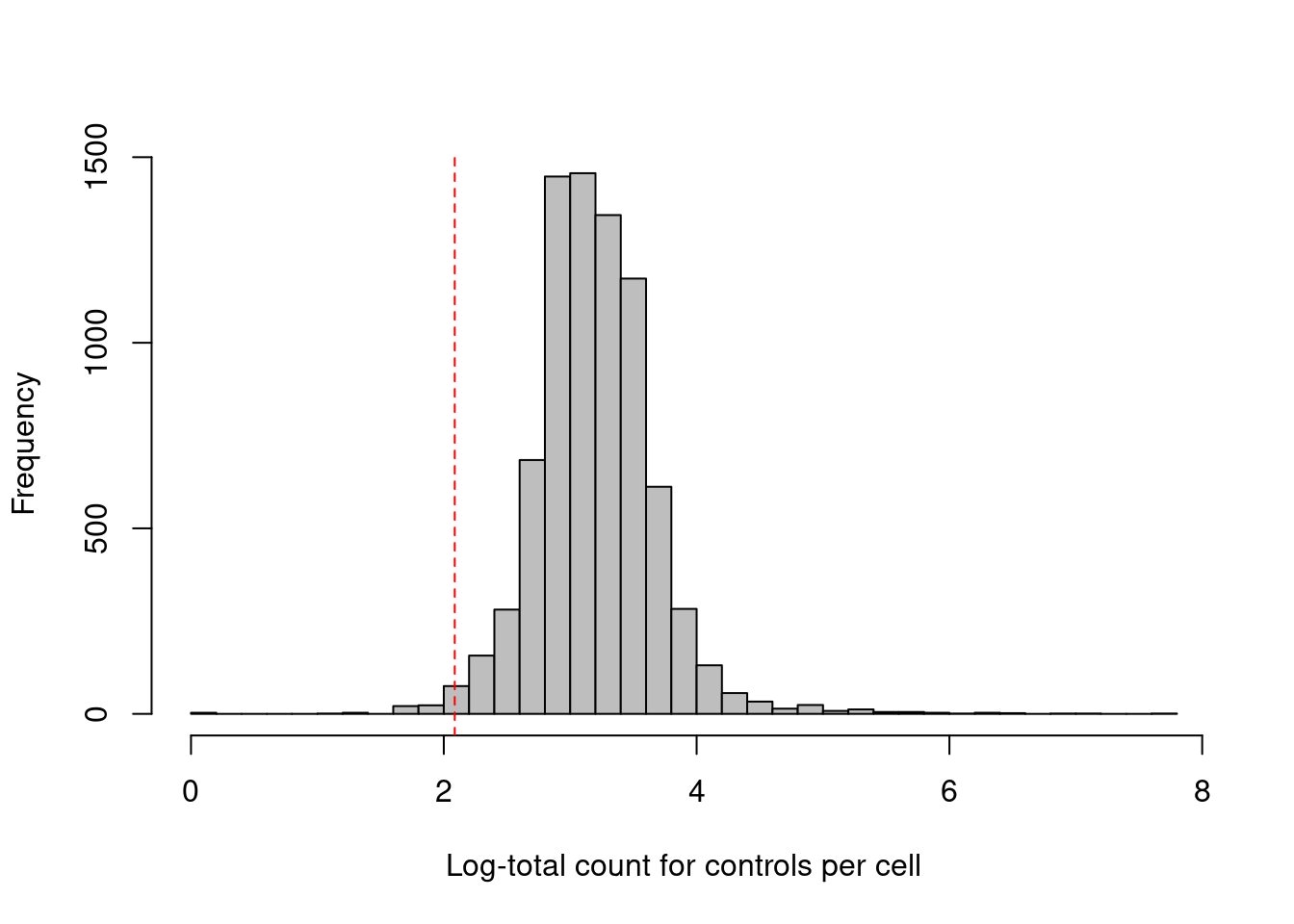
Some experiments include isotype control (IgG) antibodies that have similar properties to a primary antibody but lack a specific target in the cell, thus providing a measure of non-specific binding.
If we assume that the magnitude of non-specific binding is constant across cells, we could define low-quality cells as those with unusually low coverage of the control ADTs, presumably due to failed capture or sequencing.
We use the outlier-based approach described in Chapter \@ref(quality-control) to choose an appropriate filter threshold (Figure \@ref(fig:cite-total-con-hist));
hard thresholds are more difficult to specify due to experiment-by-experiment variation in the expected coverage of ADTs.
Paradoxically, though, this QC metric becomes less useful in well-executed experiments where there is not enough non-specific binding to obtain meaningful coverage of the control ADTs.
```r
# We could have specified subsets= in the above perCellQCMetrics call, but it's
# easier to explain one concept at a time, so we'll just repeat it here:
controls <- grep("^Ig", rownames(altExp(sce)))
df.ab2 <- perCellQCMetrics(altExp(sce), subsets=list(controls=controls))
con.discard <- isOutlier(df.ab2$subsets_controls_sum, log=TRUE, type="lower")
summary(con.discard)
```
```
## Mode FALSE TRUE
## logical 7785 80
```
```r
hist(log1p(df.ab2$subsets_controls_sum), col='grey', breaks=50,
main="", xlab="Log-total count for controls per cell")
abline(v=log1p(attr(con.discard, "thresholds")["lower"]), col="red", lty=2)
```
(\#fig:cite-total-con-hist)Distribution of the log-coverage of IgG control ADTs across all cells in the PBMC dataset. The red dotted line indicates the threshold below which cells were removed.
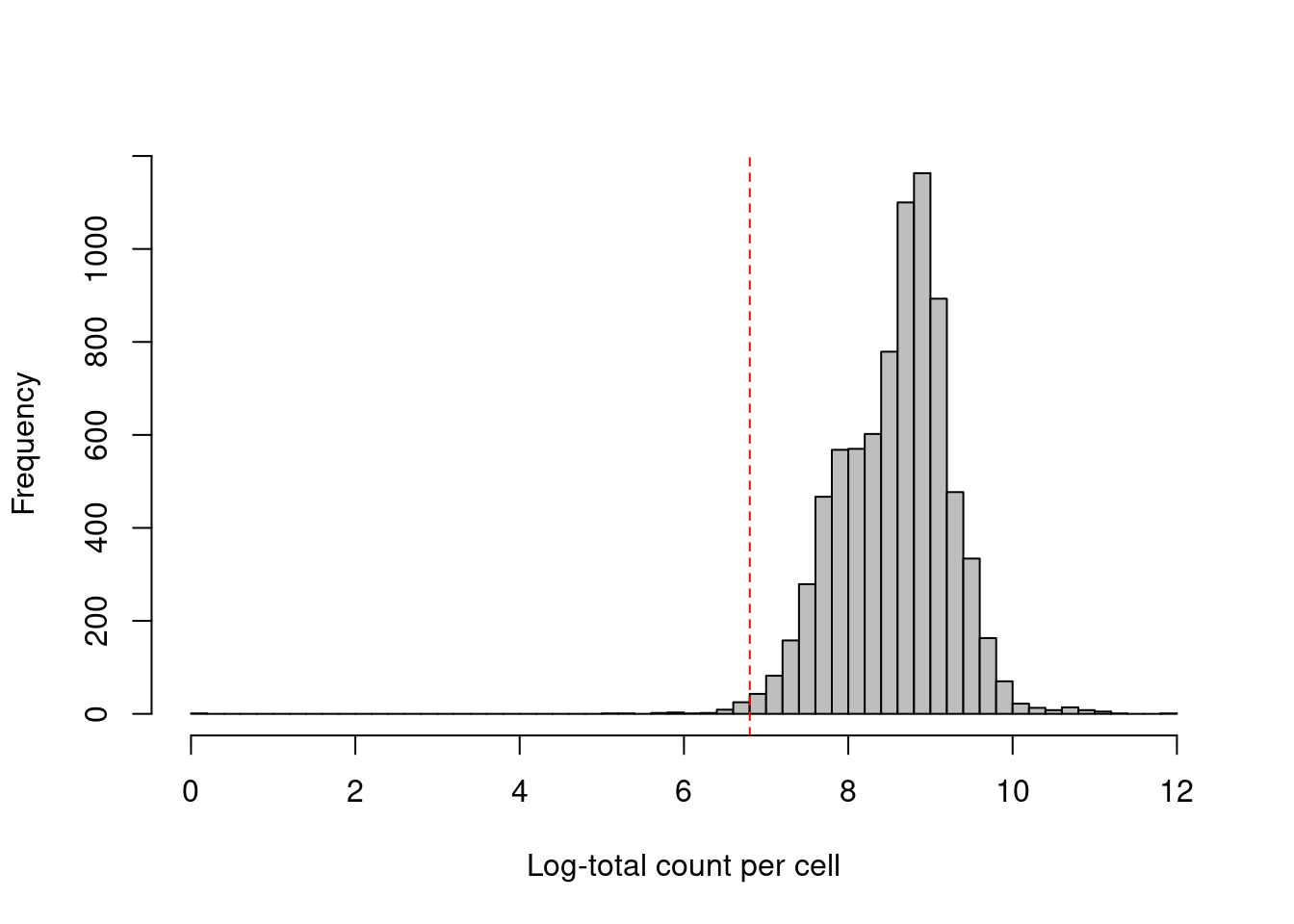
As with scRNA-seq, the total count across all ADTs can also be used as a QC metric (Figure \@ref(fig:cite-total-all-hist)).
However, it is a riskier proportion for CITE-seq as there is a greater risk that the total count will be strongly correlated with the biological state of the cell.
The presence of a targeted protein can lead to a several-fold increase in the total ADT count given the binary nature of most surface protein markers.
Removing cells with low totals could inadvertently eliminate cell types that do not express many - or indeed, any - of the selected protein targets.
Admittedly, this problem is the same as that discussed previously for the library size in scRNA-seq data (Section \@ref(outlier-assumptions)) but can be more pronounced in CITE-seq where there is no guaranteed set of constitutively expressed features to "buffer" cell-type-specific variation in the total count.
```r
sum.discard <- isOutlier(df.ab2$sum, log=TRUE, type="lower")
summary(sum.discard)
```
```
## Mode FALSE TRUE
## logical 7820 45
```
```r
hist(log1p(df.ab2$sum), col='grey', breaks=50, main="", xlab="Log-total count per cell")
abline(v=log1p(attr(sum.discard, "thresholds")["lower"]), col="red", lty=2)
```
(\#fig:cite-total-all-hist)Distribution of the log-total count of all ADTs across all cells in the PBMC dataset. The red dotted line indicates the threshold below which cells are considered to be low outliers.
Of course, if we want to analyze gene expression and ADT data together, we still need to apply quality control on the gene counts.
It is possible for a cell to have satisfactory ADT counts but poor QC metrics from the endogenous genes - for example, cell damage manifesting as high mitochondrial proportions would not be captured in the ADT data.
More generally, the ADTs are synthetic, external to the cell and conjugated to antibodies, so it is not surprising that they would experience different cell-specific technical effects than the endogenous transcripts.
This motivates a distinct QC step on the genes to ensure that we only retain high-quality cells in both feature spaces.
Here, the count matrix has already been filtered by _Cellranger_ to remove empty droplets so we only filter on the mitochondrial proportions to remove putative low-quality cells.
```r
mito <- grep("^MT-", rowData(sce)$Symbol)
df <- perCellQCMetrics(sce, subsets=list(Mito=mito))
mito.discard <- isOutlier(df$subsets_Mito_percent, type="higher")
summary(mito.discard)
```
```
## Mode FALSE TRUE
## logical 7569 296
```
Finally, to remove the low-quality cells, we subset the `SingleCellExperiment` as previously described.
This automatically applies the filtering to both the transcript and ADT data; such coordination is one of the advantages of storing both datasets in a single object.
We omit the total count filter to reduce the risk of discarding an all-negative cell type, at least for the first round of analysis.
```r
discard <- ab.discard | con.discard | mito.discard
sce <- sce[,!discard]
```
## Normalization
### Overview
Counts for the ADTs are subject to several biases that must be normalized prior to further analysis.
Capture efficiency varies from cell to cell though the differences in biophysical properties between endogenous transcripts and the (much shorter) ADTs means that the capture-related biases for the two sets of features are unlikely to be identical.
Composition biases are also much more pronounced in ADT data due to (i) the binary nature of target protein abundances, where any increase in protein abundance manifests as a large increase to the total tag count; and (ii) the _a priori_ selection of interesting protein targets, which enriches for features that are more likely to be differentially abundant across the population.
As in Chapter \@ref(normalization), we assume that these are scaling biases and compute ADT-specific size factors to remove them.
To this end, several strategies are again available to calculate a size factor for each cell.
### Library size normalization
The simplest approach is to normalize on the total ADT counts, effectively the library size for the ADTs.
Like in Section \@ref(library-size-normalization), these "ADT library size factors" are adequate for clustering but will introduce composition biases that interfere with interpretation of the fold-changes between clusters.
This is especially tue for relatively subtle (e.g., ~2-fold) changes in the abundances of markers associated with functional activity rather than cell type.
```r
sf.lib <- librarySizeFactors(altExp(sce))
summary(sf.lib)
```
```
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.031 0.528 0.906 1.000 1.266 22.672
```
We might instead consider the related approach of taking the geometric mean of all counts as the size factor for each cell [@stoeckius2017simultaneous].
The geometric mean is a reasonable estimator of the scaling biases for large counts with the added benefit that it mitigates the effects of composition biases by dampening the impact of one or two highly abundant ADTs.
While more robust than the ADT library size factors, these geometric mean-based factors are still not entirely correct and will progressively become less accurate as upregulation increases in strength.
```r
library(scuttle)
sf.geo <- geometricSizeFactors(altExp(sce))
summary(sf.geo)
```
```
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.16 0.66 0.85 1.00 1.07 45.22
```
### Median-based normalization {#cite-seq-median-norm}
Ideally, we would like to compute size factors that adjust for the composition biases.
This usually requires an assumption that most ADTs are not differentially expressed between cell types/states.
At first glance, this appears to be a strong assumption - the target proteins were specifically chosen as they exhibit interesting heterogeneity across the population, meaning that a non-differential majority across ADTs would be unlikely.
However, we can make it work by assuming that each cell only upregulates a minority of the targeted proteins, with the remaining ADTs exhibiting some low baseline abundance that is constant across all cells.
(Of course, the identity of the upregulated subset can differ across cells, otherwise the dataset would not be very interesting.)
We then compute size factors to equalize the coverage of the non-upregulated majority, thus eliminating cell-to-cell differences in capture efficiency.
We consider the baseline ADT profile to be a combination of weak constitutive expression and ambient contamination, both of which should be constant across the population.
We estimate this profile by assuming that the distribution of abundances for each ADT should be bimodal, where one population of cells exhibits low baseline expression and another population upregulates the corresponding protein target.
We then use all cells in the lower mode to compute the baseline abundance for that ADT.
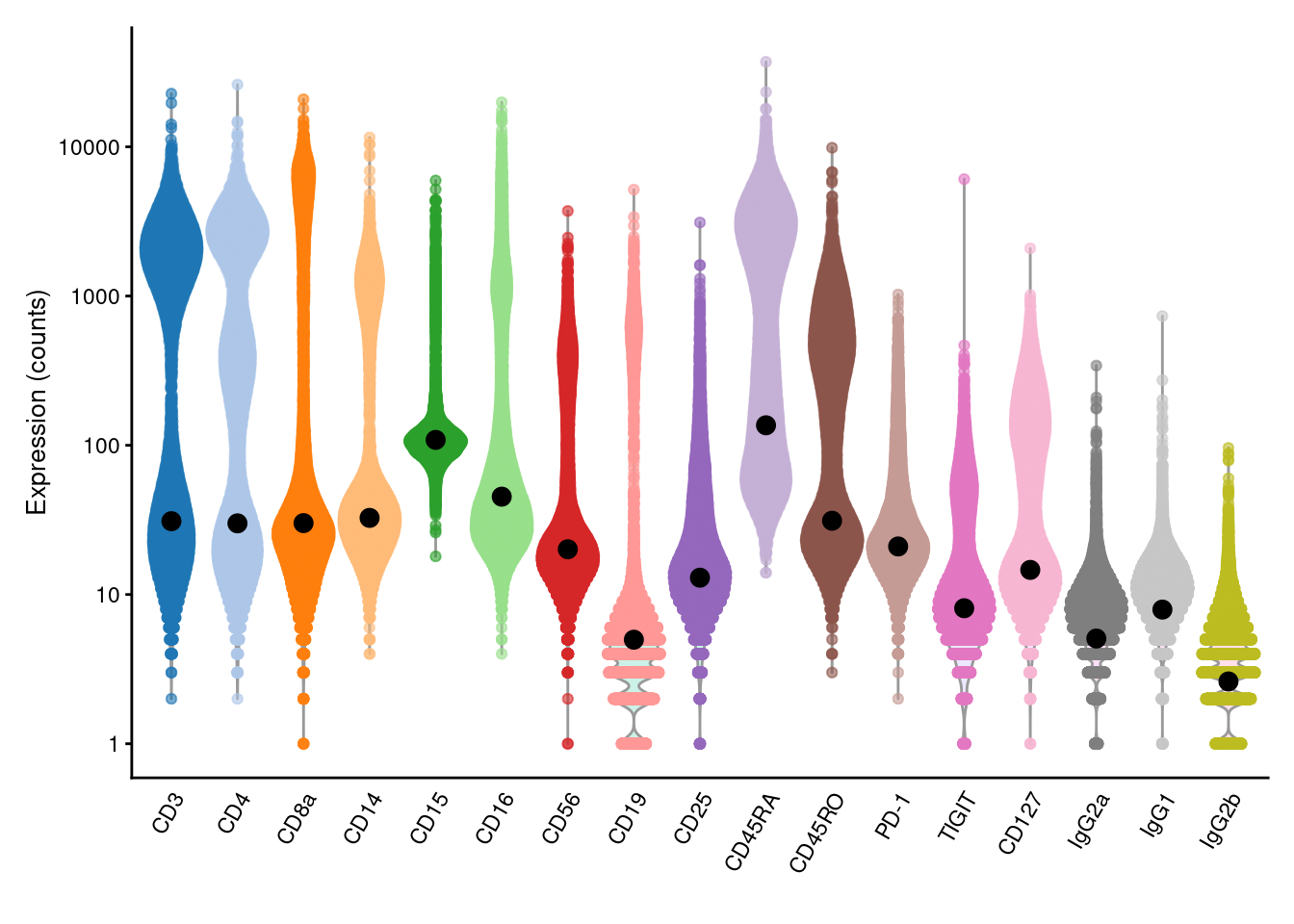
This entire calculation is performed by the `inferAmbience()` function, which was originally designed to estimate HTO ambient concentrations but can be repurposed for more general use (Figure \@ref(fig:pbmc-adt-ambient-profile)).
```r
baseline <- inferAmbience(counts(altExp(sce)))
head(baseline)
```
```
## CD3 CD4 CD8a CD14 CD15 CD16
## 30.99 29.97 30.09 32.61 108.65 45.25
```
```r
library(scater)
plotExpression(altExp(sce), features=rownames(altExp(sce)), exprs_values="counts") +
scale_y_log10() +
geom_point(data=data.frame(x=names(baseline), y=baseline), mapping=aes(x=x, y=y), cex=3)
```
(\#fig:pbmc-adt-ambient-profile)Distribution of (log-)counts for each ADT in the PBMC dataset, with the inferred ambient abundance marked by the black dot.
We use a *[DESeq2](https://bioconductor.org/packages/3.12/DESeq2)*-like approach to compute size factors against the baseline profile.
Specifically, the size factor for each cell is defined as the median of the ratios of that cell's counts to the baseline profile.
If the abundances for most ADTs in each cell are baseline-derived, they should be roughly constant across cells;
any systematic differences in the ratios correspond to cell-specific biases in sequencing coverage and are captured by the size factor.
The use of the median protects against the minority of ADTs corresponding to genuinely expressed targets.
```r
sf.amb <- medianSizeFactors(altExp(sce), reference=baseline)
summary(sf.amb)
```
```
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.20 0.66 0.78 1.00 1.00 71.00
```
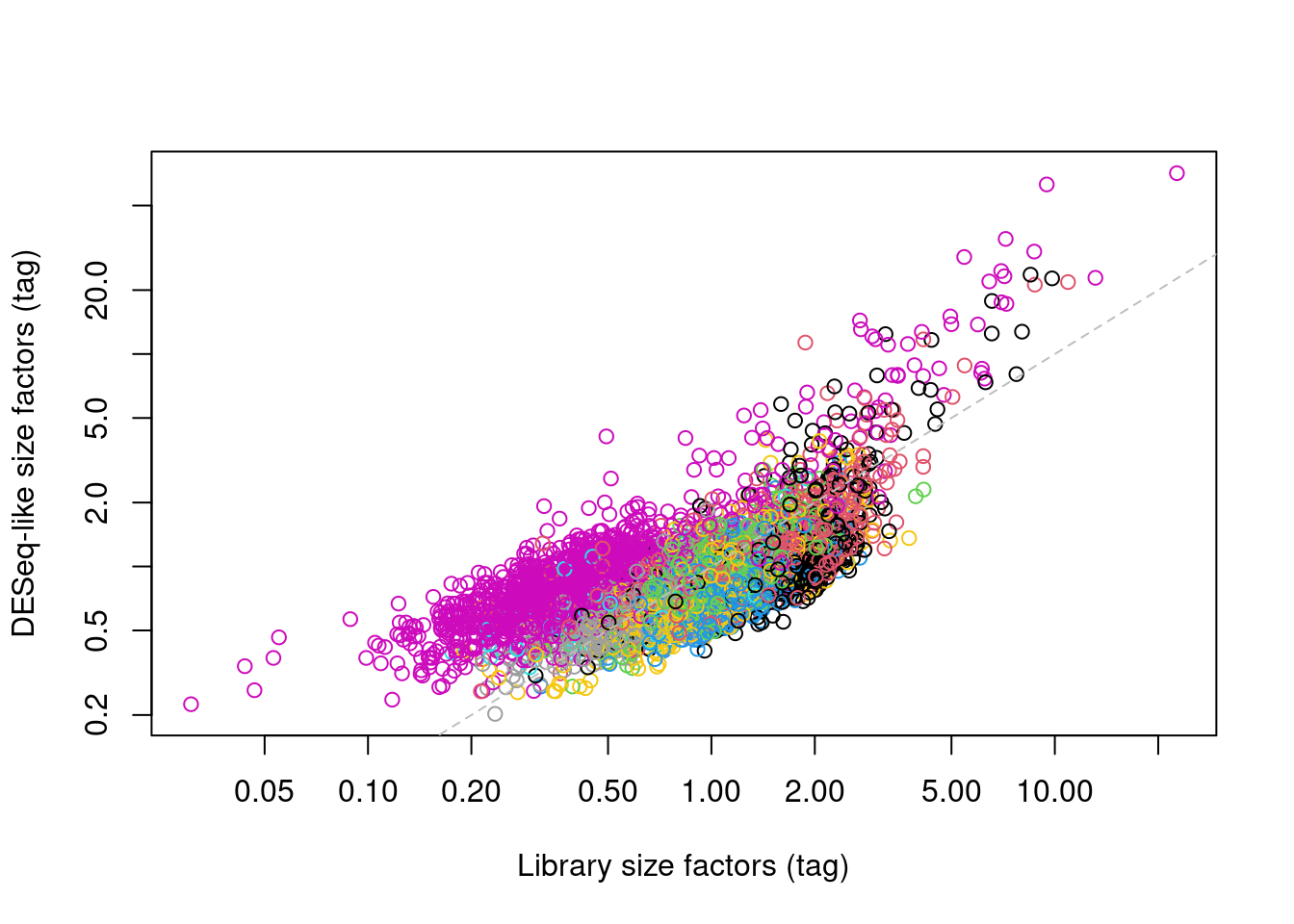
In one subpopulation, the size factors are consistently larger than the ADT library size factors, whereas the opposite is true for most of the other subpopulations (Figure \@ref(fig:comp-bias-norm)).
This is consistent with the presence of composition biases due to differential abundance of the targeted proteins between subpopulations.
Here, composition biases would introduce a spurious 2-fold change in normalized ADT abundance if the library size factors were used.
```r
# Coloring by cluster to highlight the composition biases.
# We set k=20 to get fewer, broader clusters for a clearer picture.
library(scran)
tagdata <- logNormCounts(altExp(sce)) # library size factors by default.
g <- buildSNNGraph(tagdata, k=20, d=NA) # no need for PCA, see below.
clusters <- igraph::cluster_walktrap(g)$membership
plot(sf.lib, sf.amb, log="xy", col=clusters,
xlab="Library size factors (tag)",
ylab="DESeq-like size factors (tag)")
abline(0, 1, col="grey", lty=2)
```
(\#fig:comp-bias-norm)DESeq-like size factors for each cell in the PBMC dataset, compared to ADT library size factors. Each point is a cell and is colored according to the cluster identity defined from normalized ADT data.
### Control-based normalization
If control ADTs are available, we could make the assumption that they should not be differentially abundant between cells.
Any difference thus represents some bias that should be normalized by defining control-based size factors from the sum of counts over all control ADTs, analogous to spike-in normalization (Section \@ref(spike-norm)),
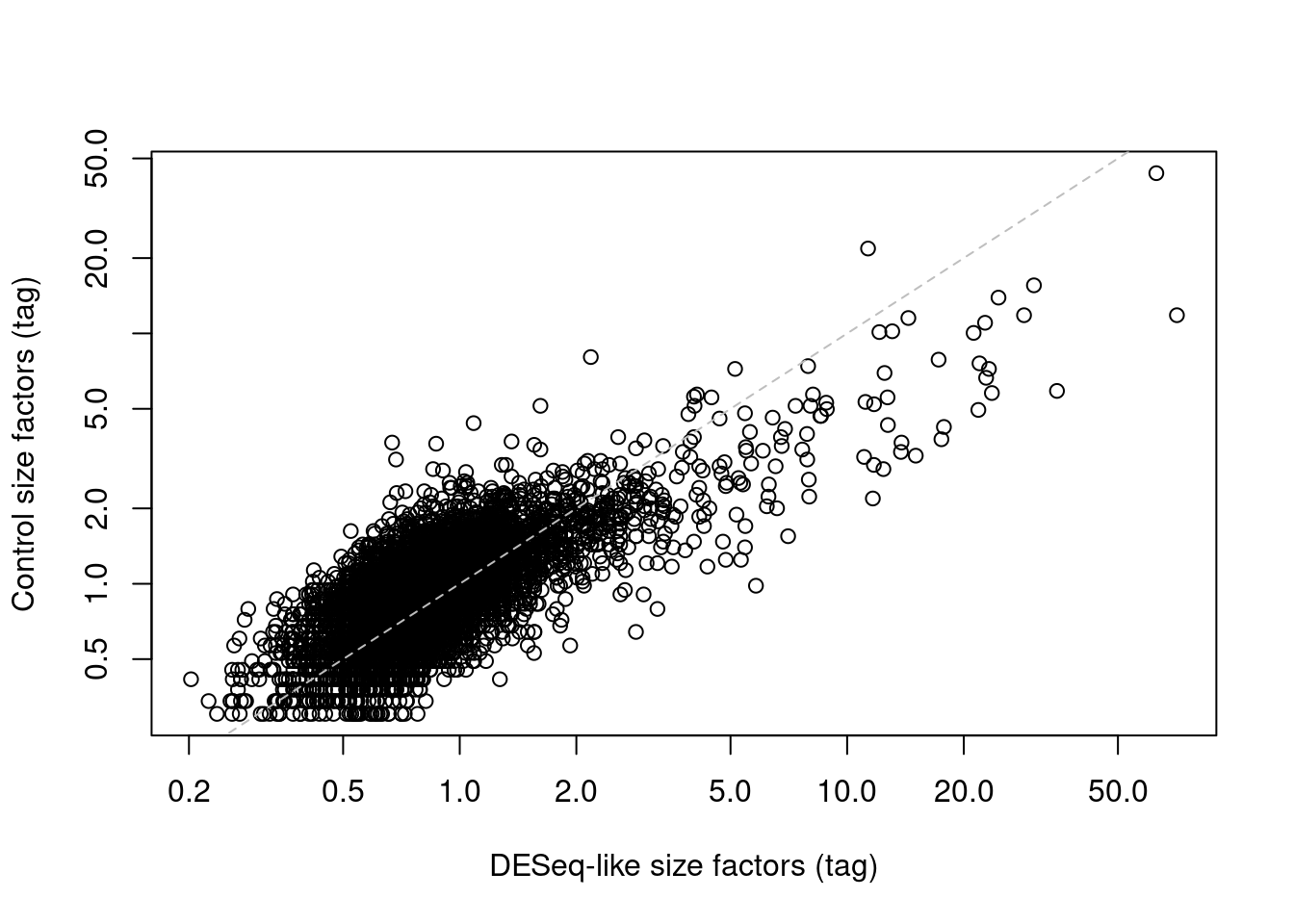
We demonstrate this approach below by computing size factors from the IgG controls (Figure \@ref(fig:control-bias-norm)).
```r
controls <- grep("^Ig", rownames(altExp(sce)))
sf.control <- librarySizeFactors(altExp(sce), subset_row=controls)
summary(sf.control)
```
```
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.30 0.68 0.87 1.00 1.13 43.67
```
```r
plot(sf.amb, sf.control, log="xy",
xlab="DESeq-like size factors (tag)",
ylab="Control size factors (tag)")
abline(0, 1, col="grey", lty=2)
```
(\#fig:control-bias-norm)IgG control-derived size factors for each cell in the PBMC dataset, compared to the DESeq-like size factors.
This approach exchanges the previous assumption of a non-differential majority for another assumption about the lack of differential abundance in the control tags.
We might feel that the latter is a generally weaker assumption, but it is possible for non-specific binding to vary due to biology (e.g., when the cell surface area increases), at which point this normalization strategy may not be appropriate.
It also relies on sufficient coverage of the control ADTs, which is not always possible in well-executed experiments where there is little non-specific binding to provide such counts.
### Computing log-normalized values
We suggest using the median-based size factors by default, as they are generally applicable and eliminate most problems with composition biases.
We set the size factors for the ADT data by calling `sizeFactors()` on the relevant `altExp()`.
In contrast, `sizeFactors(sce)` refers to size factors for the gene counts, which are usually quite different.
```r
sizeFactors(altExp(sce)) <- sf.amb
```
Regardless of which size factors are chosen, running `logNormCounts()` will then perform scaling normalization and log-transformation for both the endogenous transcripts and the ADTs using their respective size factors.
```r
sce <- logNormCounts(sce, use.altexps=TRUE)
# Checking that we have normalized values:
assayNames(sce)
```
```
## [1] "counts" "logcounts"
```
```r
assayNames(altExp(sce))
```
```
## [1] "counts" "logcounts"
```
## Clustering and interpretation
Unlike transcript-based counts, feature selection is largely unnecessary for analyzing ADT data.
This is because feature selection has already occurred during experimental design where the manual choice of target proteins means that all ADTs correspond to interesting features by definition.
From a practical perspective, the ADT count matrix is already small so there is no need for data compaction from using HVGs or PCs.
Moreover, each ADT is often chosen to capture some orthogonal biological signal, so there is not much extraneous noise in higher dimensions that can be readily removed.
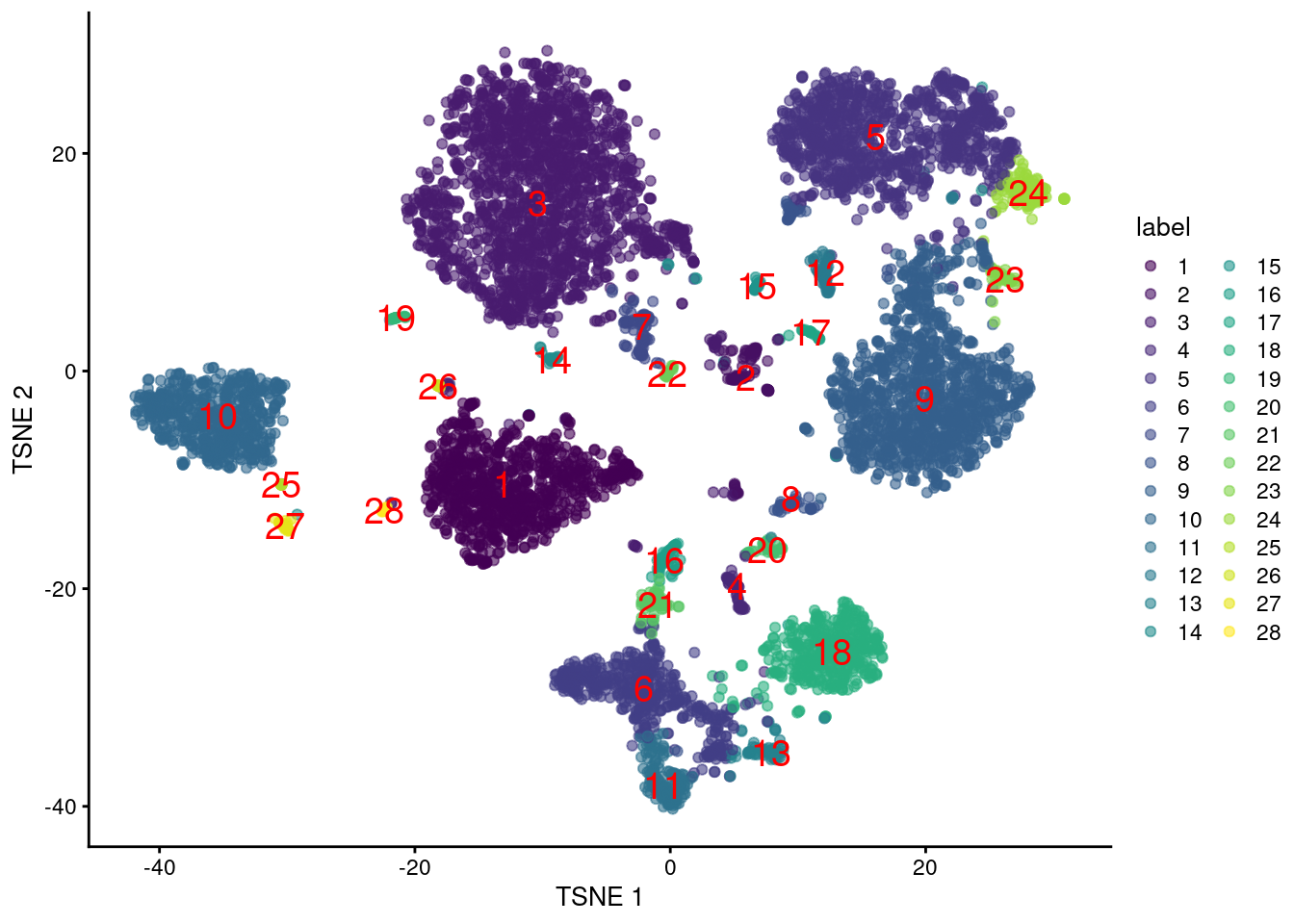
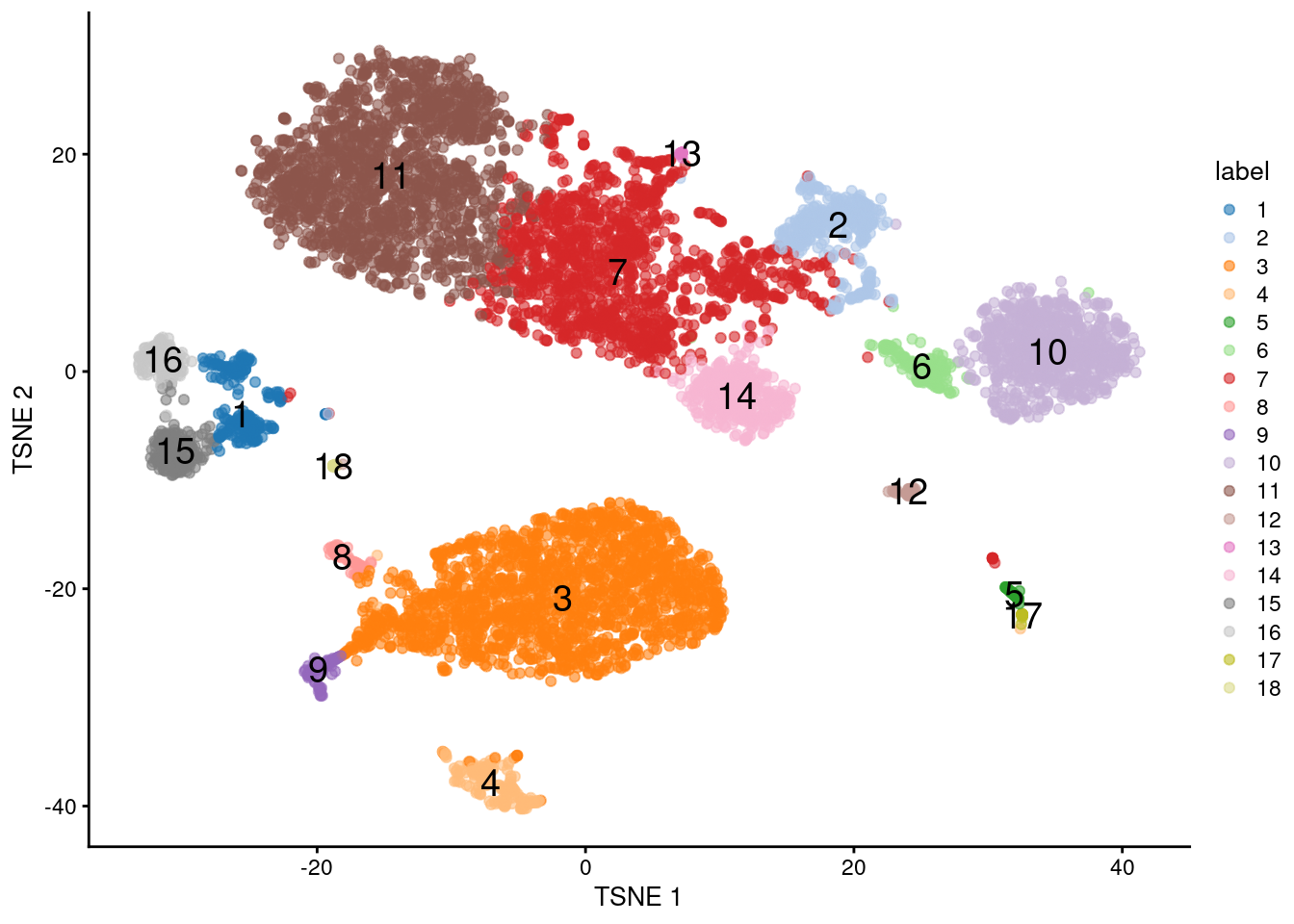
This suggests we should directly apply downstream procedures like clustering and visualization on the log-normalized abundance matrix for the ADTs (Figure \@ref(fig:tsne-tags)).
```r
# Set d=NA so that the function does not perform PCA.
g.adt <- buildSNNGraph(altExp(sce), d=NA)
clusters.adt <- igraph::cluster_walktrap(g.adt)$membership
# Generating a t-SNE plot.
library(scater)
set.seed(1010010)
altExp(sce) <- runTSNE(altExp(sce))
colLabels(altExp(sce)) <- factor(clusters.adt)
plotTSNE(altExp(sce), colour_by="label", text_by="label", text_col="red")
```
(\#fig:tsne-tags)$t$-SNE plot generated from the log-normalized abundance of each ADT in the PBMC dataset. Each point is a cell and is labelled according to its assigned cluster.
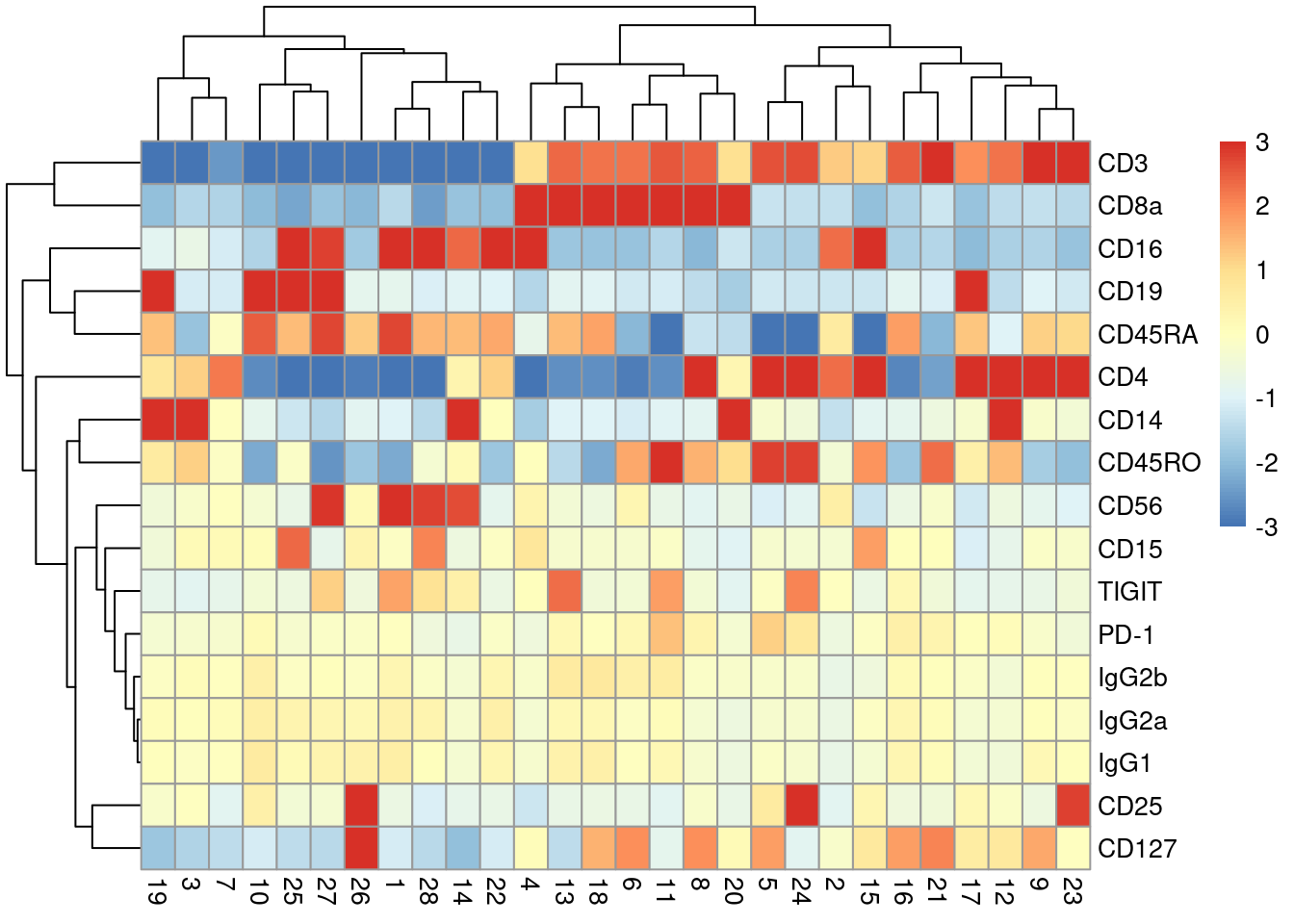
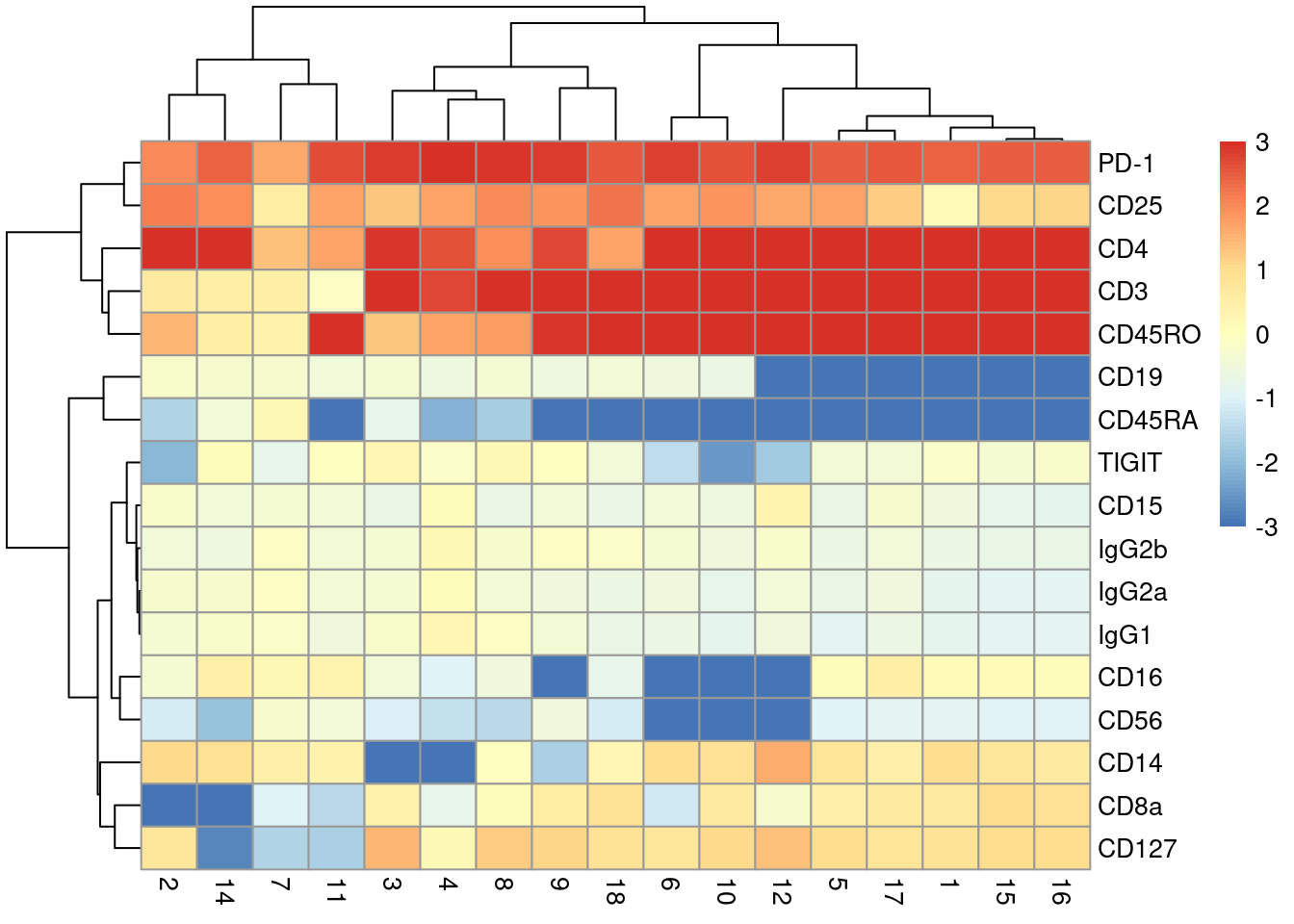
With only a few ADTs, characterization of each cluster is most efficiently achieved by creating a heatmap of the average log-abundance of each tag (Figure \@ref(fig:heat-tags)).
For this experiment, we can easily identify B cells (CD19^+^), various subsets of T cells (CD3^+^, CD4^+^, CD8^+^), monocytes and macrophages (CD14^+^, CD16^+^), to name a few.
More detailed examination of the distribution of abundances within each cluster is easily performed with `plotExpression()` where strong bimodality may indicate that finer clustering is required to resolve cell subtypes.
```r
se.averaged <- sumCountsAcrossCells(altExp(sce), clusters.adt,
exprs_values="logcounts", average=TRUE)
library(pheatmap)
averaged <- assay(se.averaged)
pheatmap(averaged - rowMeans(averaged),
breaks=seq(-3, 3, length.out=101))
```
(\#fig:heat-tags)Heatmap of the average log-normalized abundance of each ADT in each cluster of the PBMC dataset. Colors represent the log~2~-fold change from the grand average across all clusters.
Of course, this provides little information beyond what we could have obtained from a mass cytometry experiment; the real value of this data lies in the integration of protein abundance with gene expression.
## Integration with gene expression data
### By subclustering
In the simplest approach to integration, we take cells in each of the ADT-derived clusters and perform subclustering using the transcript data.
This is an _in silico_ equivalent to an experiment that performs FACS to isolate cell types followed by scRNA-seq for further characterization.
We exploit the fact that the ADT abundances are cleaner (larger counts, stronger signal) for more robust identification of broad cell types, and use the gene expression data to identify more subtle structure that manifests in the transcriptome.
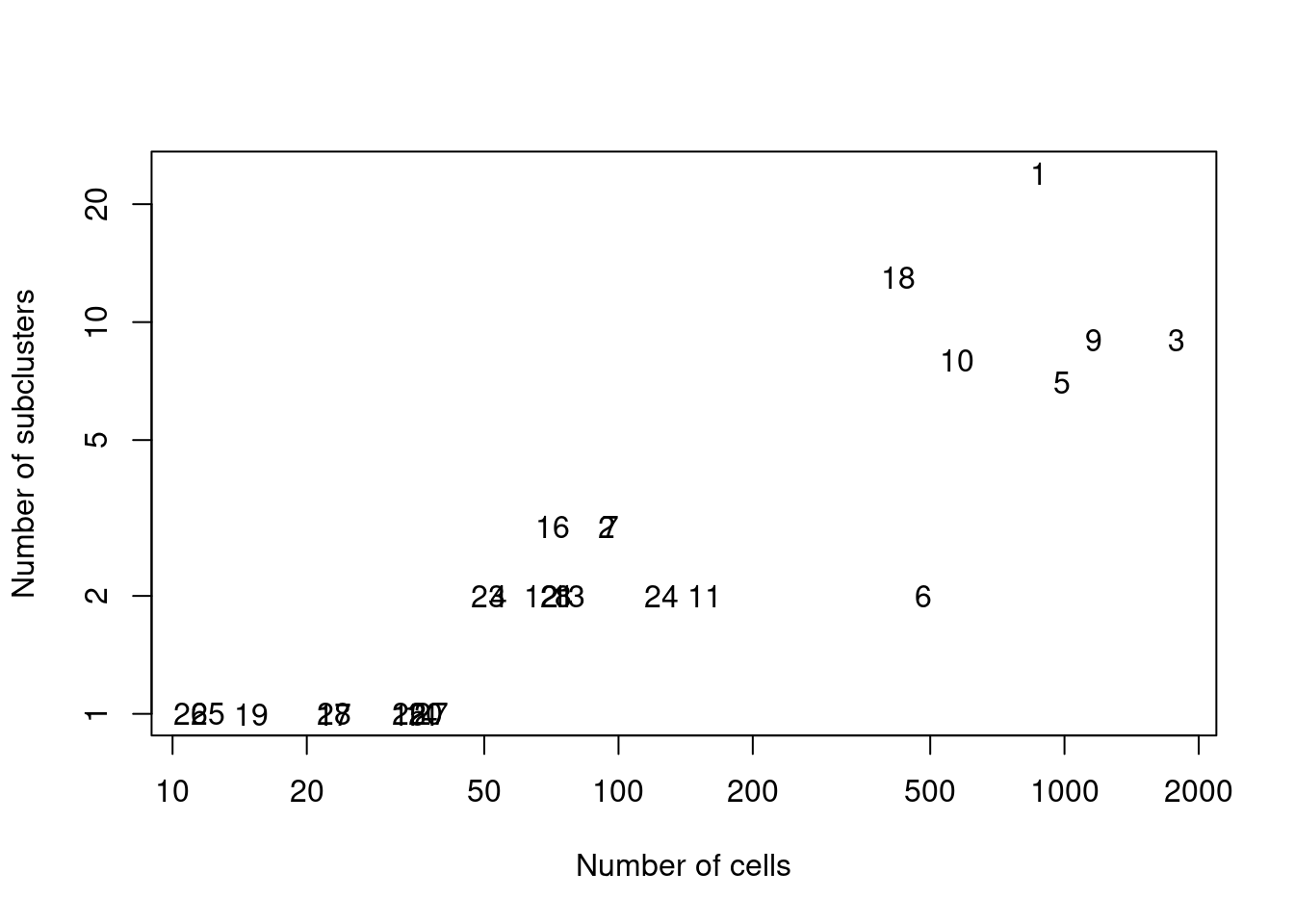
We demonstrate below by using `quickSubCluster()` to loop over all of the ADT-derived clusters and subcluster on gene expression (Figure \@ref(fig:subcluster-stats)).
```r
set.seed(101010)
all.sce <- quickSubCluster(sce, clusters.adt,
prepFUN=function(x) {
dec <- modelGeneVar(x)
top <- getTopHVGs(dec, prop=0.1)
x <- runPCA(x, subset_row=top, ncomponents=25)
},
clusterFUN=function(x) {
g.trans <- buildSNNGraph(x, use.dimred="PCA")
igraph::cluster_walktrap(g.trans)$membership
}
)
# Summarizing the number of subclusters in each tag-derived parent cluster,
# compared to the number of cells in that parent cluster.
ncells <- vapply(all.sce, ncol, 0L)
nsubclusters <- vapply(all.sce, FUN=function(x) length(unique(x$subcluster)), 0L)
plot(ncells, nsubclusters, xlab="Number of cells", type="n",
ylab="Number of subclusters", log="xy")
text(ncells, nsubclusters, names(all.sce))
```
(\#fig:subcluster-stats)Number of subclusters identified from the gene expression data within each ADT-derived parent cluster.
Another benefit of subclustering is that we can use the annotation on the ADT-derived clusters to facilitate annotation of each subcluster.
If we knew that cluster `X` contained T cells from the ADT-derived data, there is no need to identify subclusters `X.1`, `X.2`, etc. as T cells from scratch; rather, we can focus on the more subtle (and interesting) differences between the subclusters using `findMarkers()`.
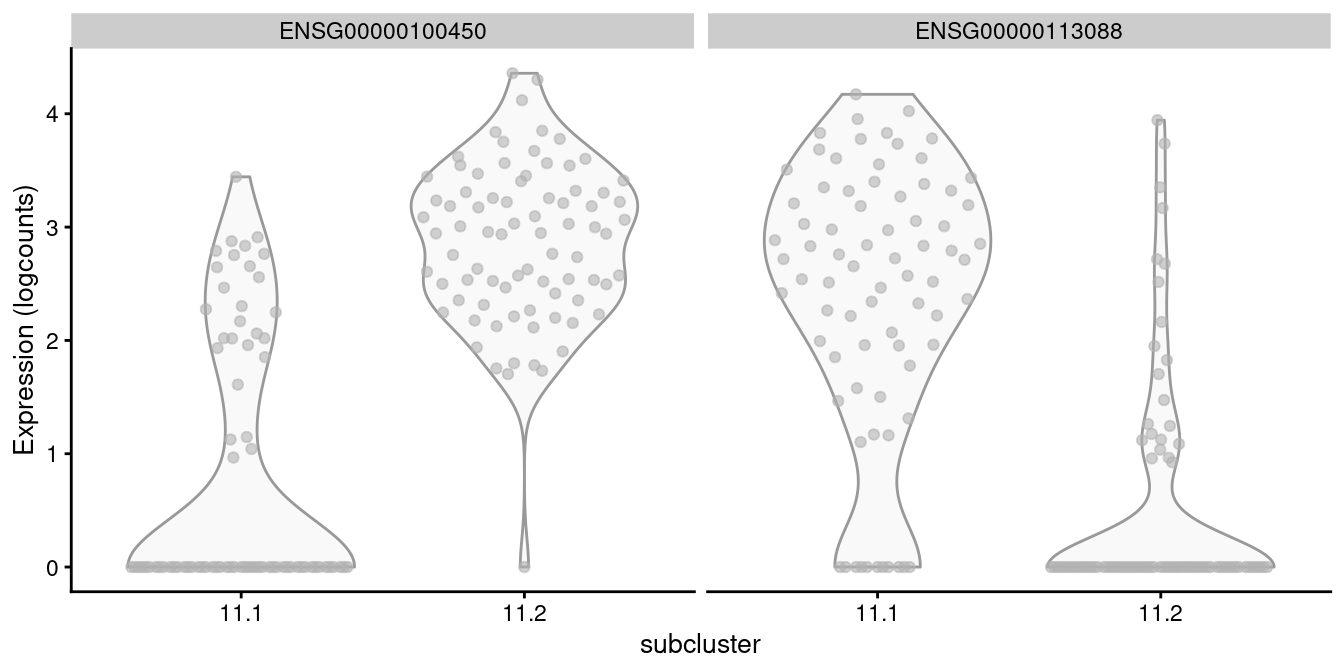
For example, cluster 11 contains CD8^+^ T cells according to Figure \@ref(fig:heat-tags), in which we further identify internal subclusters based on granzyme expression (Figure \@ref(fig:gzmh-cd8-t)).
Subclustering is also conceptually appealing as it avoids comparing log-fold changes in protein abundances with log-fold changes in gene expression.
This ensures that variation (or noise) from the transcript counts does not compromise cell type/state identification from the relatively cleaner ADT counts.
```r
of.interest <- "11"
plotExpression(all.sce[[of.interest]], x="subcluster",
features=c("ENSG00000100450", "ENSG00000113088"))
```
(\#fig:gzmh-cd8-t)Distribution of log-normalized expression values of _GZMH_ (left) and _GZHK_ (right) in transcript-derived subclusters of a ADT-derived subpopulation of CD8^+^ T cells.
The downside is that relying on previous results increases the risk of misleading conclusions when ambiguities in those results are not considered, as previously discussed in Section \@ref(subclustering).
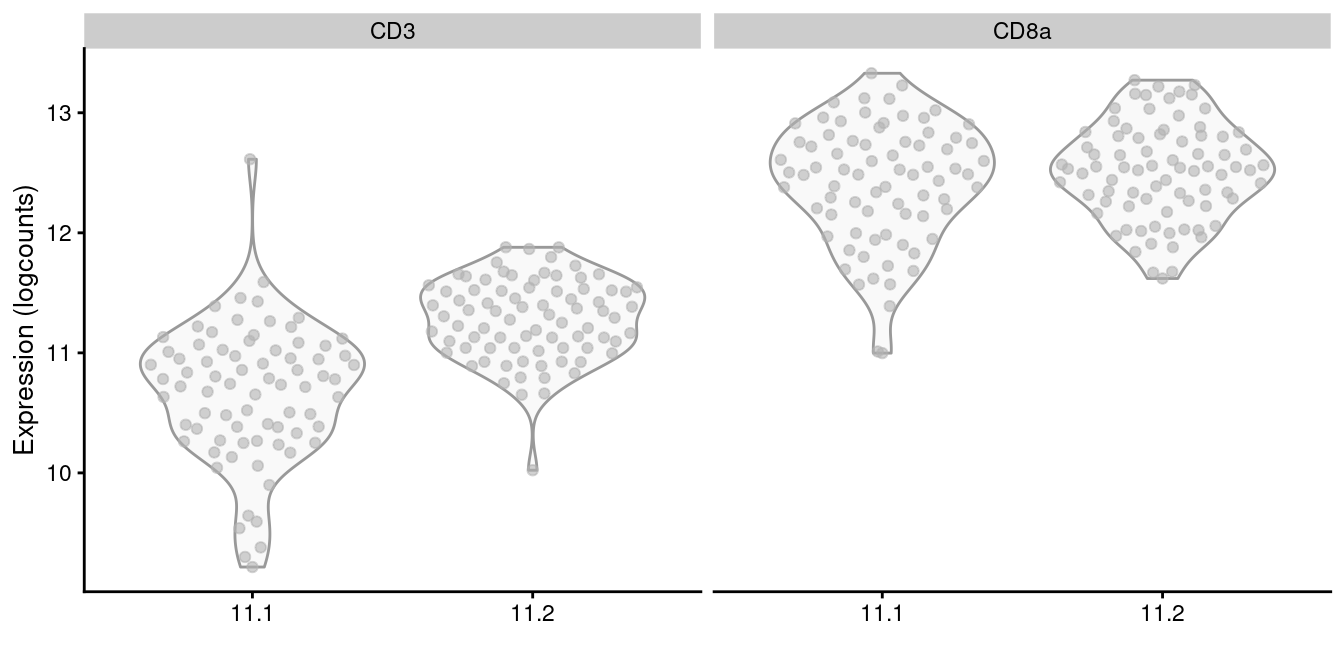
It is a good idea to perform some additional checks to ensure that each subcluster has similar protein abundances, e.g., using a heatmap as in Figure \@ref(fig:heat-tags) or with a series of plots like in Figure \@ref(fig:subcluster-tag-dist).
If so, this allows the subcluster to "inherit" the annotation attached to the parent cluster for easier interpretation.
```r
sce.cd8 <- all.sce[[of.interest]]
plotExpression(altExp(sce.cd8), x=I(sce.cd8$subcluster),
features=c("CD3", "CD8a"))
```
(\#fig:subcluster-tag-dist)Distribution of log-normalized abundances of ADTs for CD3 and CD8a in each subcluster of the CD8^+^ T cell population.
### By combined clustering
Alternatively, we can combine the information from both sets of features into a single matrix for use in downstream analyses.
This is logistically convenient as the combined structure is compatible with routine analysis workflows for transcript-only data.
To illustrate, we first perform some standard steps on the transcript count matrix:
```r
sce.main <- logNormCounts(sce)
dec.main <- modelGeneVar(sce.main)
top.main <- getTopHVGs(dec.main, prop=0.1)
sce.main <- runPCA(sce.main, subset_row=top.main, ncomponents=25)
```
The simplest version of this idea involves literally combining the log-normalized abundance matrix for the ADTs with the log-expression matrix (or its compacted form, the matrix of PCs) to obtain a single matrix for use in downstream procedures.
This requires some reweighting to balance the contribution of the transcript and ADT data to the total variance in the combined matrix, especially given that the former has around 100-fold more features than the latter.
We see that the number of clusters is slightly higher than that from the ADT data alone, consistent with the introduction of additional heterogeneity when the two feature sets are combined.
```r
# TODO: push this into a function somewhere.
library(DelayedMatrixStats)
transcript.data <- logcounts(sce.main)[top.main,,drop=FALSE]
transcript.var <- sum(rowVars(DelayedArray(transcript.data)))
tag.data <- logcounts(altExp(sce.main))
tag.var <- sum(rowVars(DelayedArray(tag.data)))
reweight <- sqrt(transcript.var/tag.var)
combined <- rbind(transcript.data, tag.data*reweight)
# 'buildSNNGraph' conveniently performs the PCA for us if requested. We use
# more PCs in 'd' to capture more variance in both sets of features. Note that
# this uses IRLBA by default so we need to set the seed.
set.seed(100010)
g.com <- buildSNNGraph(combined, d=50)
clusters.com <- igraph::cluster_walktrap(g.com)$membership
table(clusters.com)
```
```
## clusters.com
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
## 51 70 347 116 77 842 504 1121 1679 60 52 340 167 47 19 53
## 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32
## 71 981 40 36 16 68 25 25 388 73 35 11 67 20 47 27
## 33
## 17
```
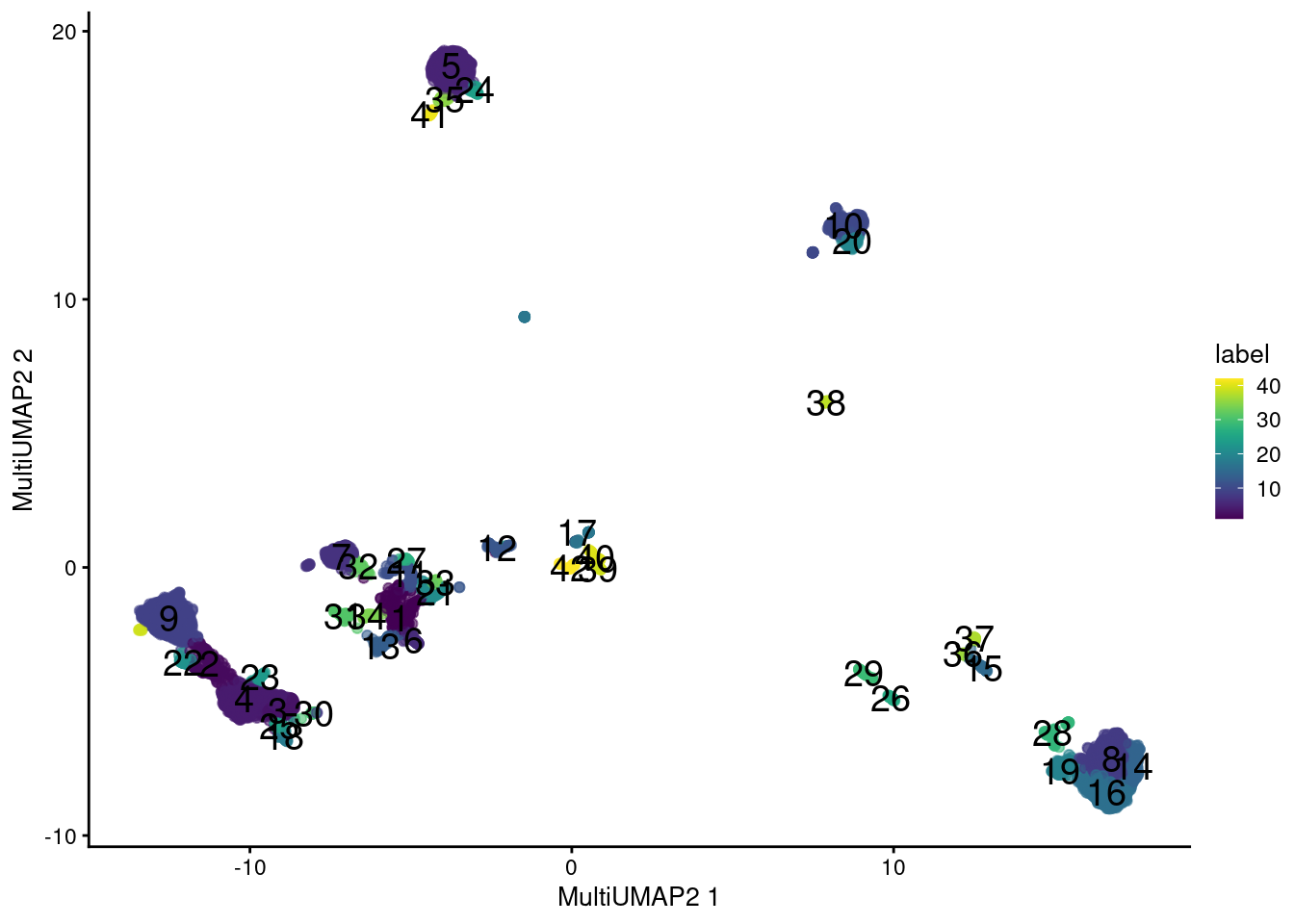
A more sophisticated approach uses the UMAP algorithm [@mcInnes2018umap] to integrate information from the two sets of features.
Very loosely speaking, we can imagine this as an intersection of the nearest neighbor graphs formed from each set, which effectively encourages the formation of communities of cells that are close in both feature spaces.
Here, we perform two rounds of UMAP; one round retains high dimensionality for a faithful representation of the data during clustering, while the other performs dimensionality reduction for a pretty visualization.
This yields an extremely fine-grained clustering in Figure \@ref(fig:combined-umap), which is attributable to the stringency of intersection operations for defining the local neighborhood.
```r
set.seed(1001010)
sce.main <- runMultiUMAP(sce.main, dimred="PCA",
altexp="Antibody Capture", n_components=50)
# Bumping up 'k' for less granularity.
g.com2 <- buildSNNGraph(sce.main, k=20, use.dimred="MultiUMAP")
clusters.com2 <- igraph::cluster_walktrap(g.com2)$membership
table(clusters.com2)
```
```
## clusters.com2
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
## 324 173 220 704 790 123 395 733 1022 426 78 67 103 181 62 763
## 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32
## 50 96 87 159 90 53 33 71 62 65 33 53 42 28 61 34
## 33 34 35 36 37 38 39 40 41 42
## 47 33 29 23 33 36 20 32 26 32
```
```r
# Combining again for visualization:
set.seed(0101110)
sce.main <- runMultiUMAP(sce.main, dimred="PCA",
altexp="Antibody Capture", name="MultiUMAP2")
colLabels(sce.main) <- clusters.com2
plotReducedDim(sce.main, "MultiUMAP2", colour_by="label", text_by="label")
```
(\#fig:combined-umap)UMAP plot obtained by combining transcript and ADT data in the PBMC dataset. Each point represents a cell and is colored according to its assigned cluster.
An even more sophisticated approach uses factor analysis to identify common and unique factors of variation in each feature set.
The set of factors can then be used as low-dimensional coordinates for each cell in downstream analyses, though a number of additional statistics are also computed that may be useful, e.g., the contribution of each feature to each factor.
```r
# Waiting for MOFA2.
```
These combined strategies are convenient but do not consider (or implicitly make assumptions about) the importance of heterogeneity in the ADT data relative to the transcript data.
For example, the UMAP approach takes equal contributions from both sets of features to the intersection, which may not be appropriate if the biology of interest is concentrated in only one set.
More generally, a combined analysis must consider the potential for uninteresting noise in one set to interfere with biological signal in the other set, a concern that is largely avoided during subclustering.
### By differential testing
In more interesting applications of this technology, protein targets are chosen that reflect some functional activity rather than cell type.
(Because, frankly, the latter is not particularly hard to infer from transcript data in most cases.)
A particularly elegant example involves quantification of the immune response by using antibodies to target the influenza peptide-MHCII complexes in T cells, albeit for mass cytometry [@fehlings2018multiplex].
If the aim is to test for differences in the functional readout, a natural analysis strategy is to use the transcript data for clustering (Figure \@ref(fig:tsne-naive)) and perform differential testing between clusters or conditions for the relevant ADTs.
```r
# Performing a quick analysis of the gene expression data.
sce <- logNormCounts(sce)
dec <- modelGeneVar(sce)
top <- getTopHVGs(dec, prop=0.1)
set.seed(1001010)
sce <- runPCA(sce, subset_row=top, ncomponents=25)
g <- buildSNNGraph(sce, use.dimred="PCA")
clusters <- igraph::cluster_walktrap(g)$membership
colLabels(sce) <- factor(clusters)
set.seed(1000010)
sce <- runTSNE(sce, dimred="PCA")
plotTSNE(sce, colour_by="label", text_by="label")
```
(\#fig:tsne-naive)$t$-SNE plot of the PBMC dataset based on the transcript data. Each point is a cell and is colored according to the assigned cluster.
We demonstrate this approach using `findMarkers()` to test for differences in tag abundance between clusters (Chapter \@ref(marker-detection)).
For example, if the PD-1 level was a readout for some interesting phenotype - say, T cell exhaustion [@pauken2015overcoming] - we might be interested in its upregulation in cluster 13 compared to all other clustuers (Figure \@ref(fig:heat-pd-1)).
Methods from Chapter \@ref(multi-sample-comparisons) can be similarly used to test for differences between conditions based on pseudo-bulk ADT counts.
```r
markers <- findMarkers(altExp(sce), colLabels(sce))
of.interest <- markers[[13]]
pheatmap(getMarkerEffects(of.interest), breaks=seq(-3, 3, length.out=101))
```
(\#fig:heat-pd-1)Heatmap of log-fold changes in tag abundances in cluster 13 compared to all other clusters identified from transcript data in the PBMC data set.
The main appeal of this approach is that it avoids data snooping (Section \@ref(from-data-snooping)) as the clusters are defined without knowledge of the ADTs.
This improves the statistical rigor of the subsequent differential testing on the ADT abundances (though only to some extent; other problems are still present, such as the lack of true replication in between-cluster comparisons).
From a practical perspective, this approach yields fewer clusters and reduces the amount of work involved in manual annotation, especially if there are multiple functional states (e.g., stressed, apoptotic, stimulated) for each cell type.
However, it is fundamentally limited to per-tag inferences; if we want to identify subpopulations with interesting combinations of target proteins, we must resort to high-dimensional analyses like clustering on the ADT abundances.
## Session Info {-}